Step 0: Upload the PDB file to the server. Since the calculations rely on the protein structure, there are few additional considerations that will ensure the reliability of the calculations and model predictions (visit here).
Step 1: The pStab web server iteratively mutates the charged (+Asn/Gln) residues limiting the number of mutations per mutant as specified by the user and calculates net electrostatic interaction energy (Eelec) (described here) at the user specified pH and ionic strength conditions.
Step 2: The server predicts the effect of mutation(s) on the protein stability using Eelec as proxy and provides details on mutational hot-spots and a list of top 5000 mutants sorted based on their net electrostatic interaction energies.
Step 3: A fundamental model parameter (described here, corresponding to the interaction energy per native contact) is tuned to reproduce experimental melting temperature (Tm) and the same set of parameters is employed to predict the mutant unfolding thermodynamics.
Step 4: The predicted thermal unfolding profile and the folding probability of the residues are made available for further analysis.
Note: Step 1 and 2 can be skippped to predict the (un)folding thermodynamics of the WT protein alone.
Please go through the detailed instructions provided below carefully to understand the allowed range of parameters and model boundaries before proceeding with the calculations.
Prepare Input Files
Follow the standard formatting used in the PDB files but ensure that the following constraints are satisfied before uploading the PDB file into the server
- The protein length should be between 30 – 300 amino acids for calculating the effect of mutations on the net charge-charge interaction energy, while 150 amino acids length is the upper bound for predicting folding thermodynamics from the WSME model.
- The PDB file should not contain any hetero atom or non-standard amino acids.
- The PDB file should contain the complete description of heavy atoms (To model missing atoms, visit here)
If the uploaded file / PDB ID contains multiple chains, only the first chain entry will be considered by the server unless otherwise mentioned. The same holds true for the NMR models, where only the first model will be considered for the further predictions. Look here for more details on errors and warnings generated by the web-server.
Upload PDB Files
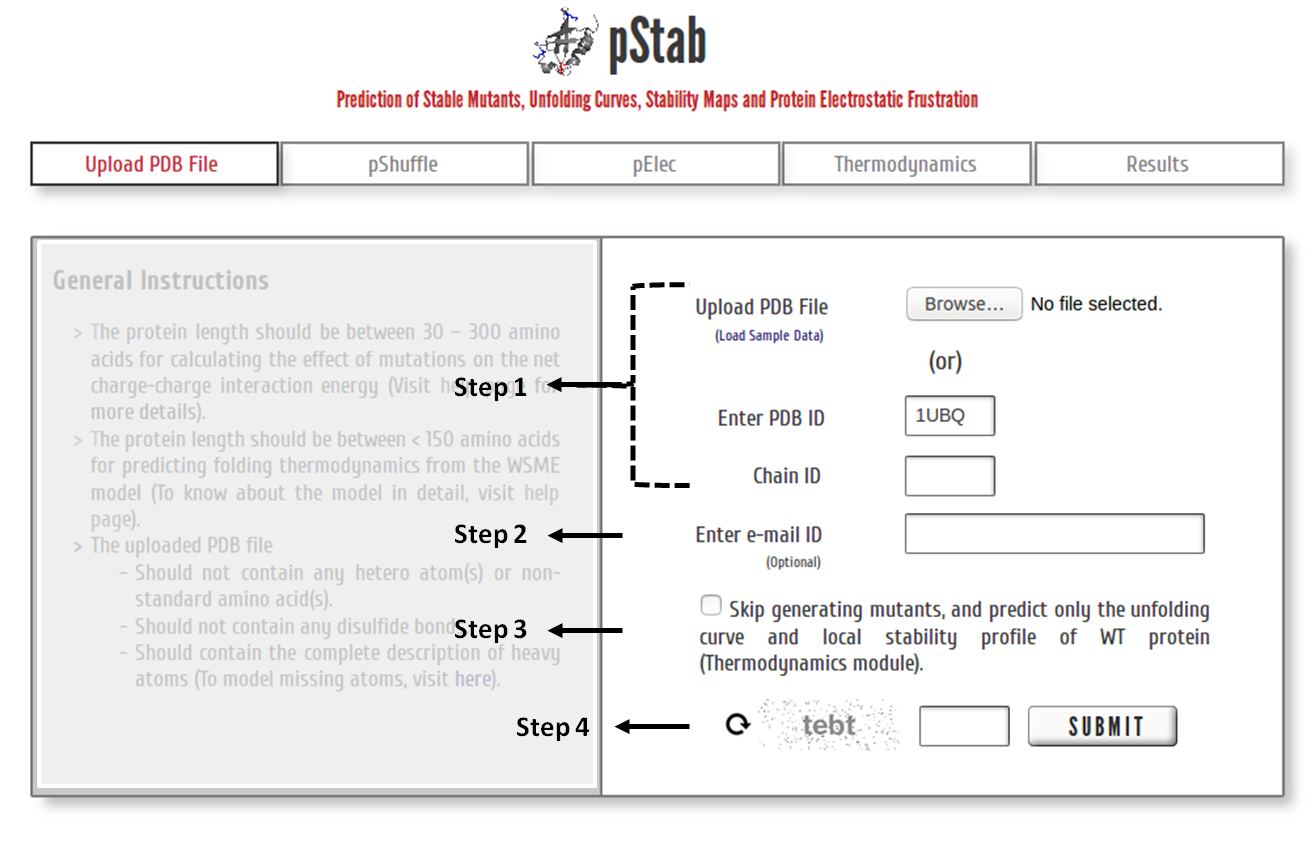 Step 1: Upload the PDB file satisfying the above-specified considerations (or) Enter a valid PDB ID. (Optional) Specify chain ID of the protein to be considered.
Step 1: Upload the PDB file satisfying the above-specified considerations (or) Enter a valid PDB ID. (Optional) Specify chain ID of the protein to be considered.
Step 2: The calculations and model predictions from the server may take a few minutes to a few hours depending on the protein size and the number of mutants to be generated (see below). You may provide your e-mail ID (optional) to which the results will be forwarded once the calculations are done.
Step 3: Click on the check box to skip the pShuffle and pElec modules and predict the (un)folding thermodynamics of the WT protein.
Step 4: Enter valid CAPTCHA and click on “SUBMIT” to proceed.
Specify Input Parameters
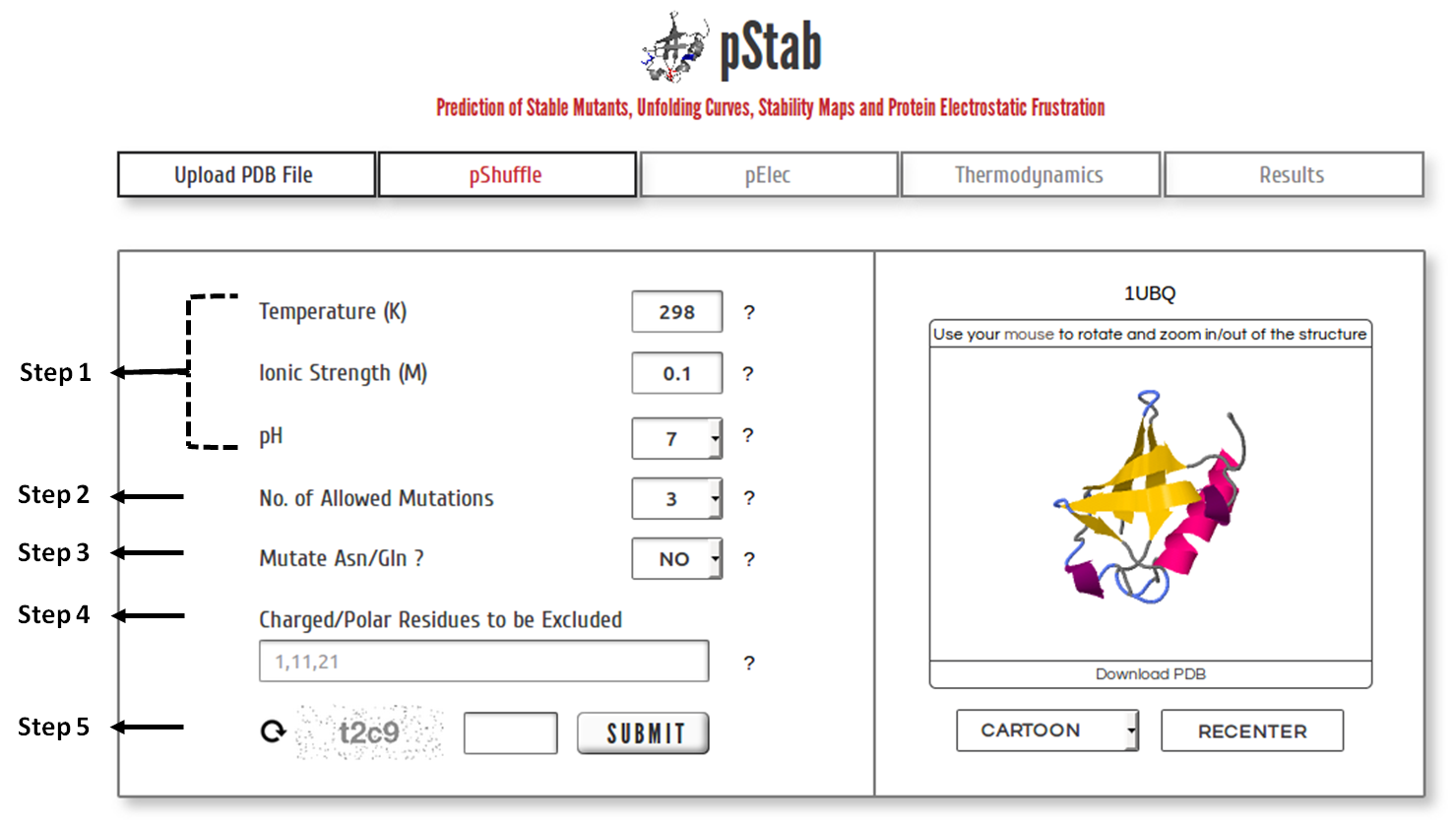 Step 1: Upload the PDB file satisfying the above-specified considerations (or) Enter a valid PDB ID. (Optional) Specify chain ID of the protein to be considered.
Step 1: Upload the PDB file satisfying the above-specified considerations (or) Enter a valid PDB ID. (Optional) Specify chain ID of the protein to be considered.
Step 2: The calculations and model predictions from the server may take a few minutes to a few hours depending on the protein size and the number of mutants to be generated (see below). You may provide your e-mail ID (optional) to which the results will be forwarded once the calculations are done.
Step 3: Click on the check box to skip the pShuffle and pElec modules and predict the (un)folding thermodynamics of the WT protein.
Step 4: Enter valid CAPTCHA and click on “SUBMIT” to proceed.
Mutational Hot-Spots and Potential Stability Effects of Mutations
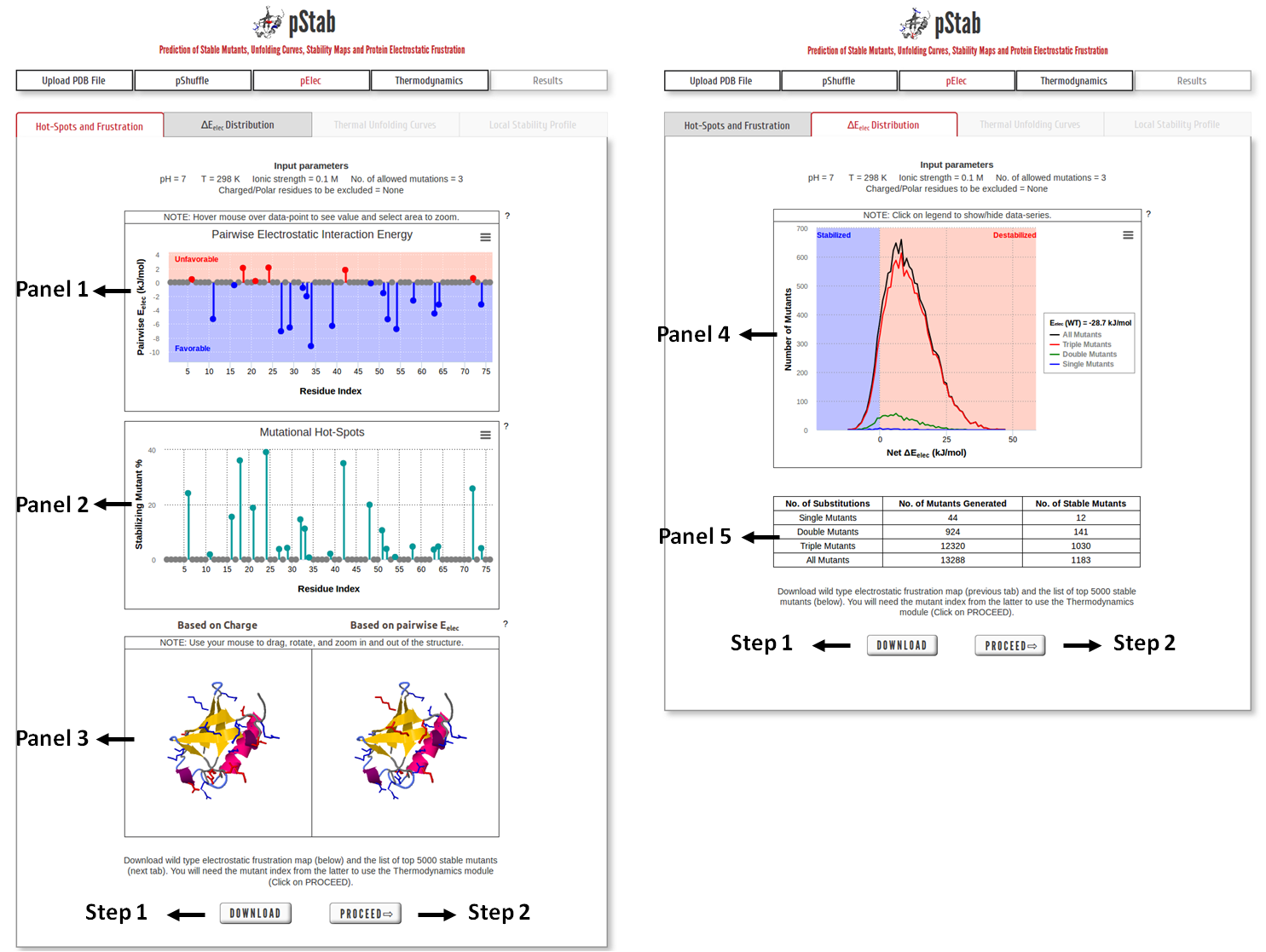
Panel 1: Pairwise Electrostatic Interaction energy as the function of residue index.
Panel 2: The graph shows the percentage of stable mutants with a mutation in that position based on the net electrostatic interaction energy criterion.
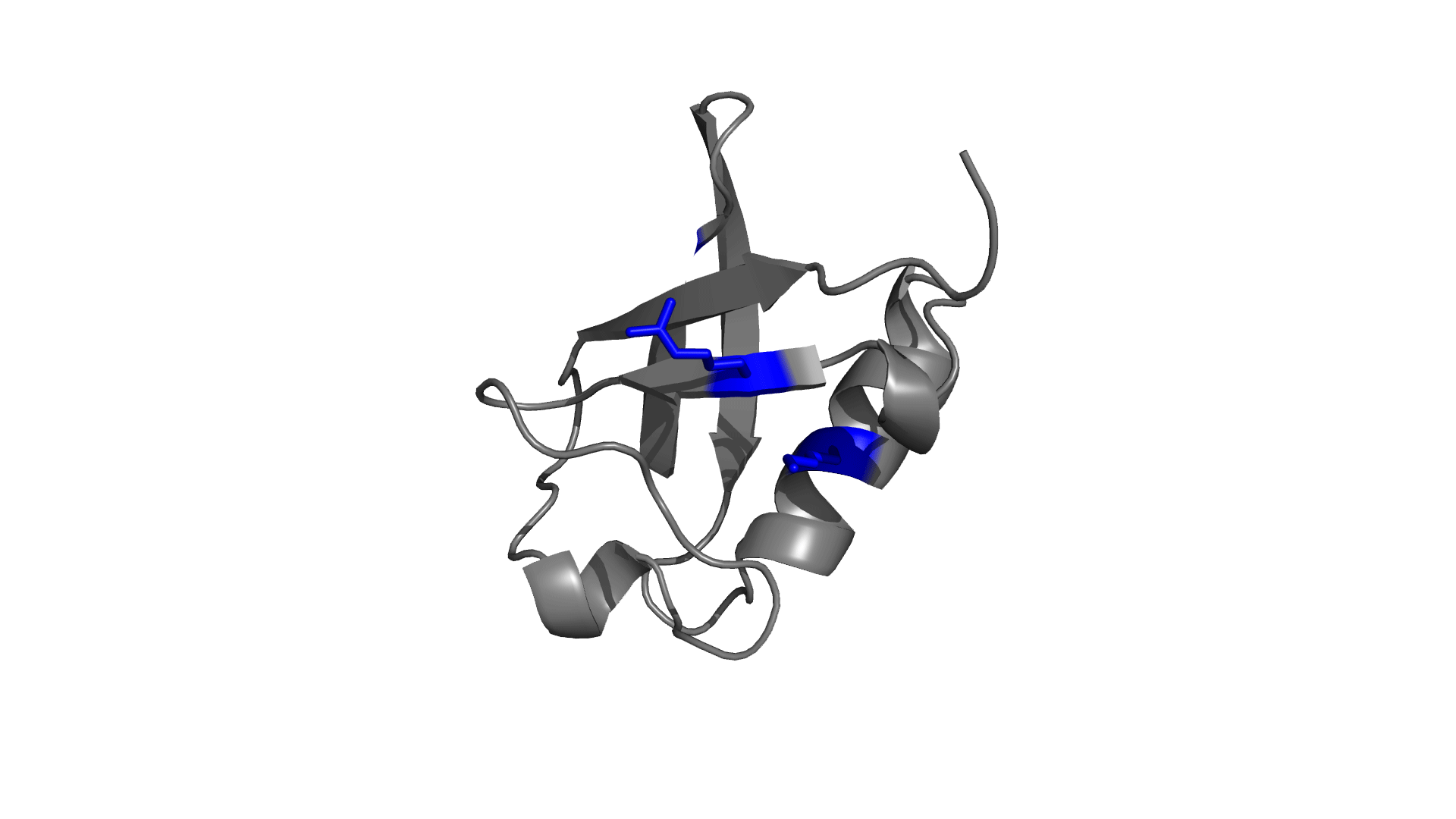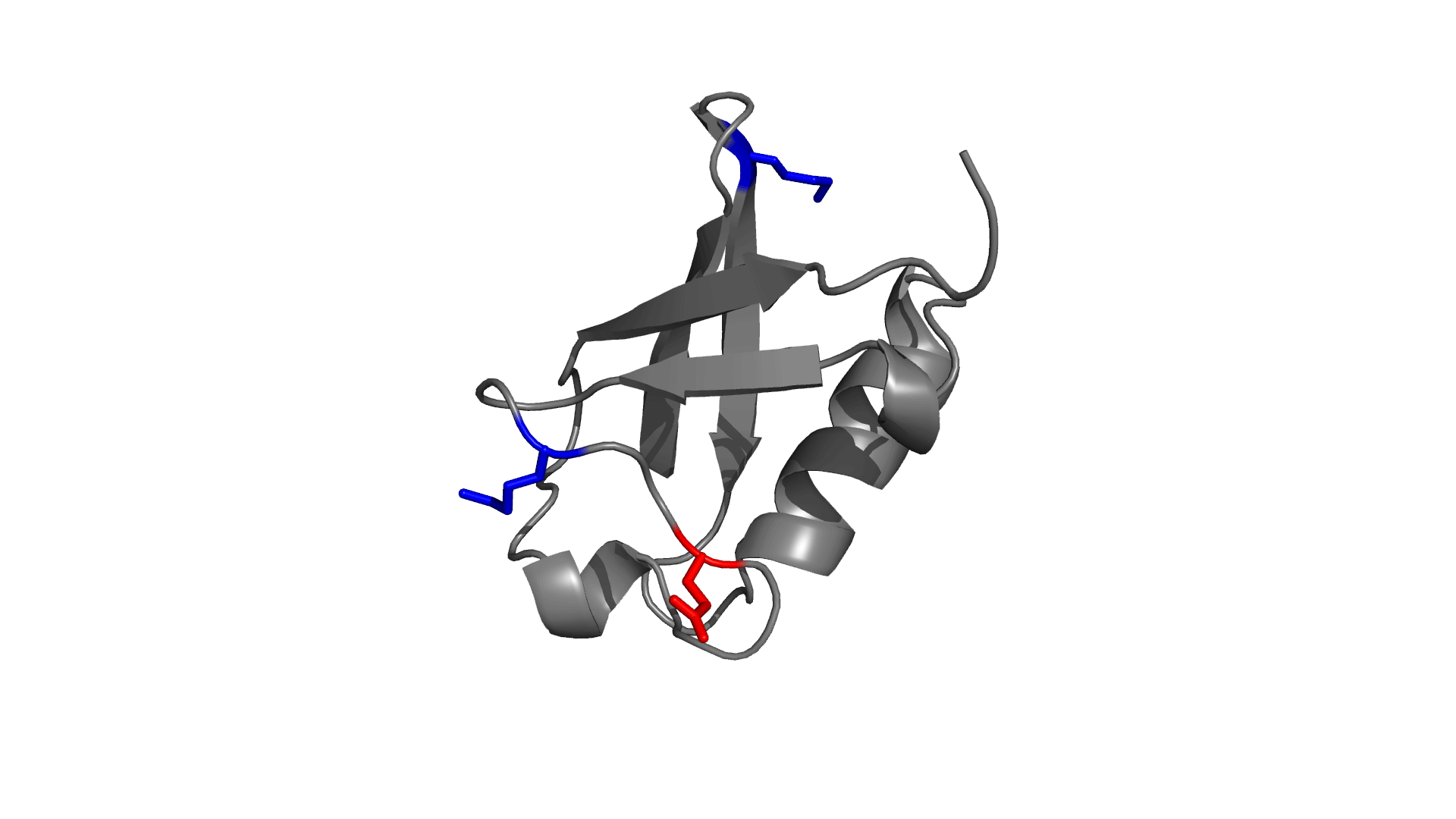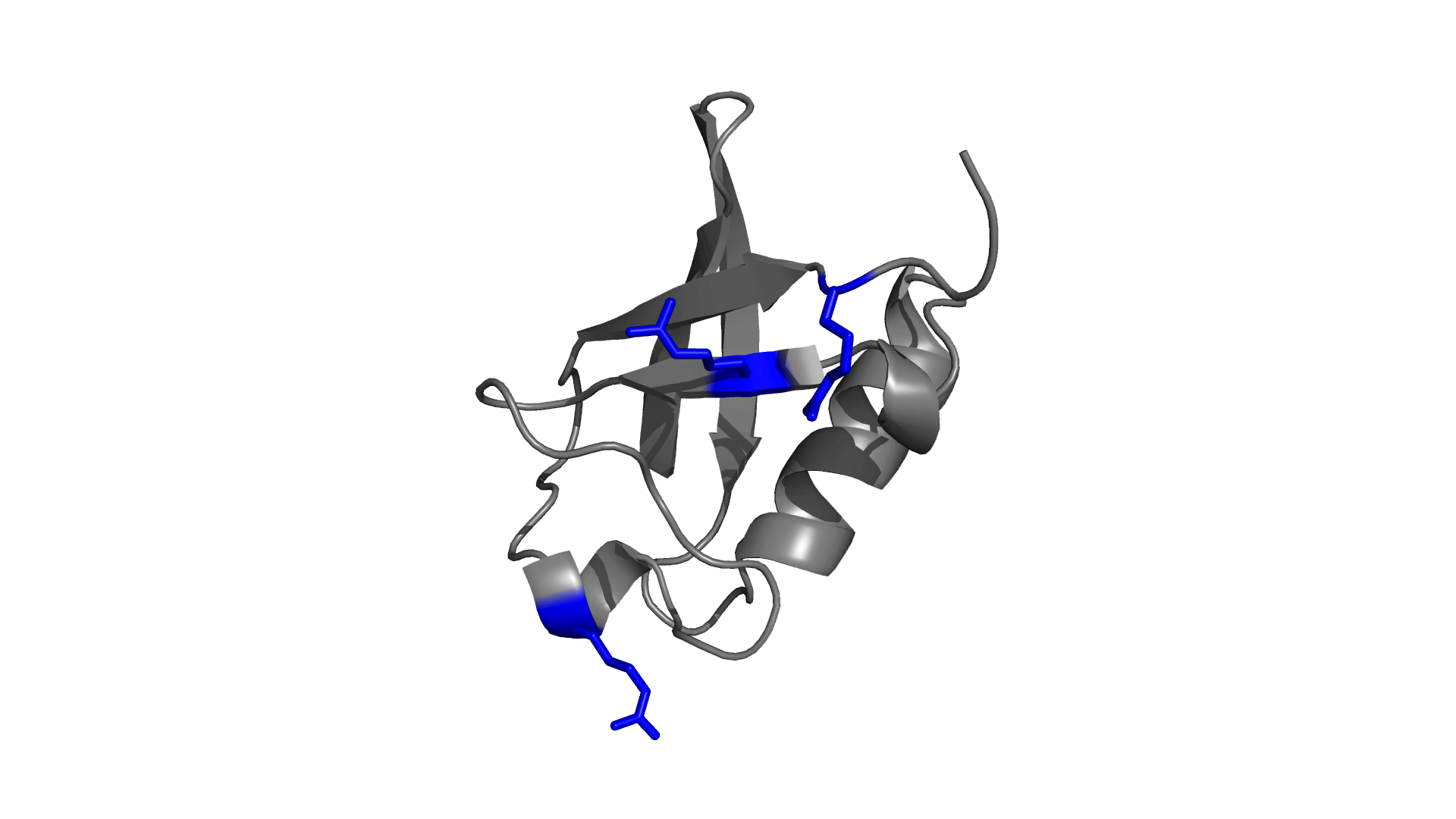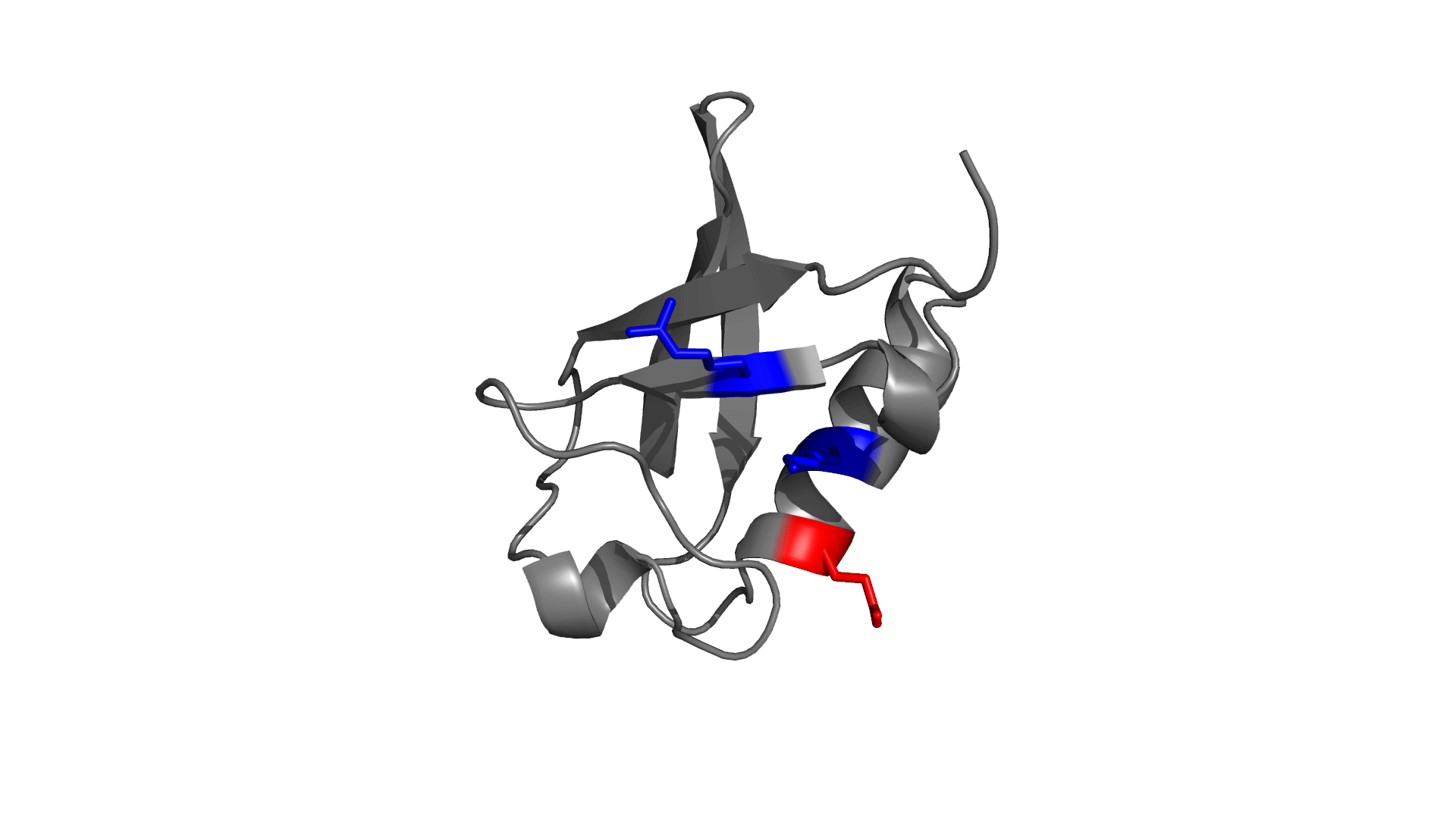
Panel 3: Visualizing the pairwise electrostatic interaction energy by mapping it on to the protein structure. In left panel, residues are colored based on their charges (red: negatively charged, blue: positively charged) and in right panel, residues are colored based on pairwise electrostatic interaction energy (red: destabilized, blue: stabilized).
Panel 4: Distribution of estimated relative stability of mutants based on ∆Eelec (=Eelec,Mut-Eelect,WT).
Panel 5: Table describing the total number of mutants generated, and the number of stabilized mutants.
Step 1: Click on “DOWNLOAD” to save the output data files into your local machine.
Step 2: Click on “PROCEED” to predict the thermodynamics of the mutants employing an ensemble based statistical-mechanical model.
Reproduce Experimental Wild-Type Protein Melting Temperature
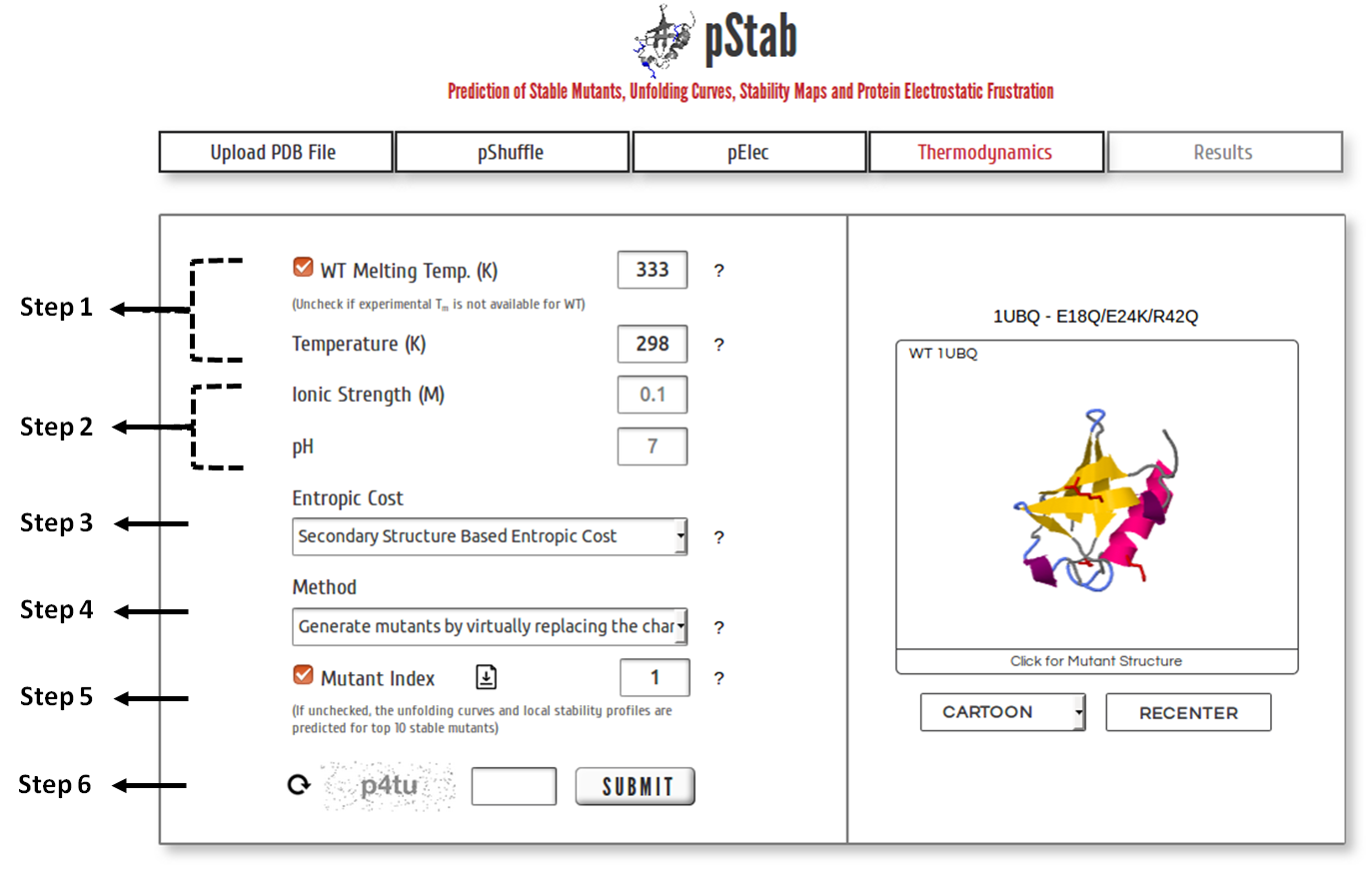 Step 1: Specify the experimentally determined melting temperature of wild type protein. Uncheck the box if experimental data is unavailable and a default value of 333 K will be used. The local stability profile of the protein(s) is predicted at the mentioned temperature.
Step 1: Specify the experimentally determined melting temperature of wild type protein. Uncheck the box if experimental data is unavailable and a default value of 333 K will be used. The local stability profile of the protein(s) is predicted at the mentioned temperature.
Step 2: The values of ionic strength and pH are restricted to the values provided initially for calculating EElec of mutants.
Step 3: Choose one of the three approaches (refer here) available to assign entropic cost to the residues.
Step 4: Choose one of the two methods available to generate mutants of interest (refer here).
Step 5: If unchecked, (un)folding thermodynamics is predicted for top 10 mutants sorted based on their stability (Refer MutantIndexFile.txt). Else, the predictions are provided for the mutant identified through the mutant index from MutantIndexFile.txt. Multiple mutant indeces separated by comma can be entered (maximum 10 entries).
Note: Step 4 and 5 are available only after the pShuffle and pElec module.
Step 6: Click on “SUBMIT” to proceed with the calculation.
Protein Thermodynamics
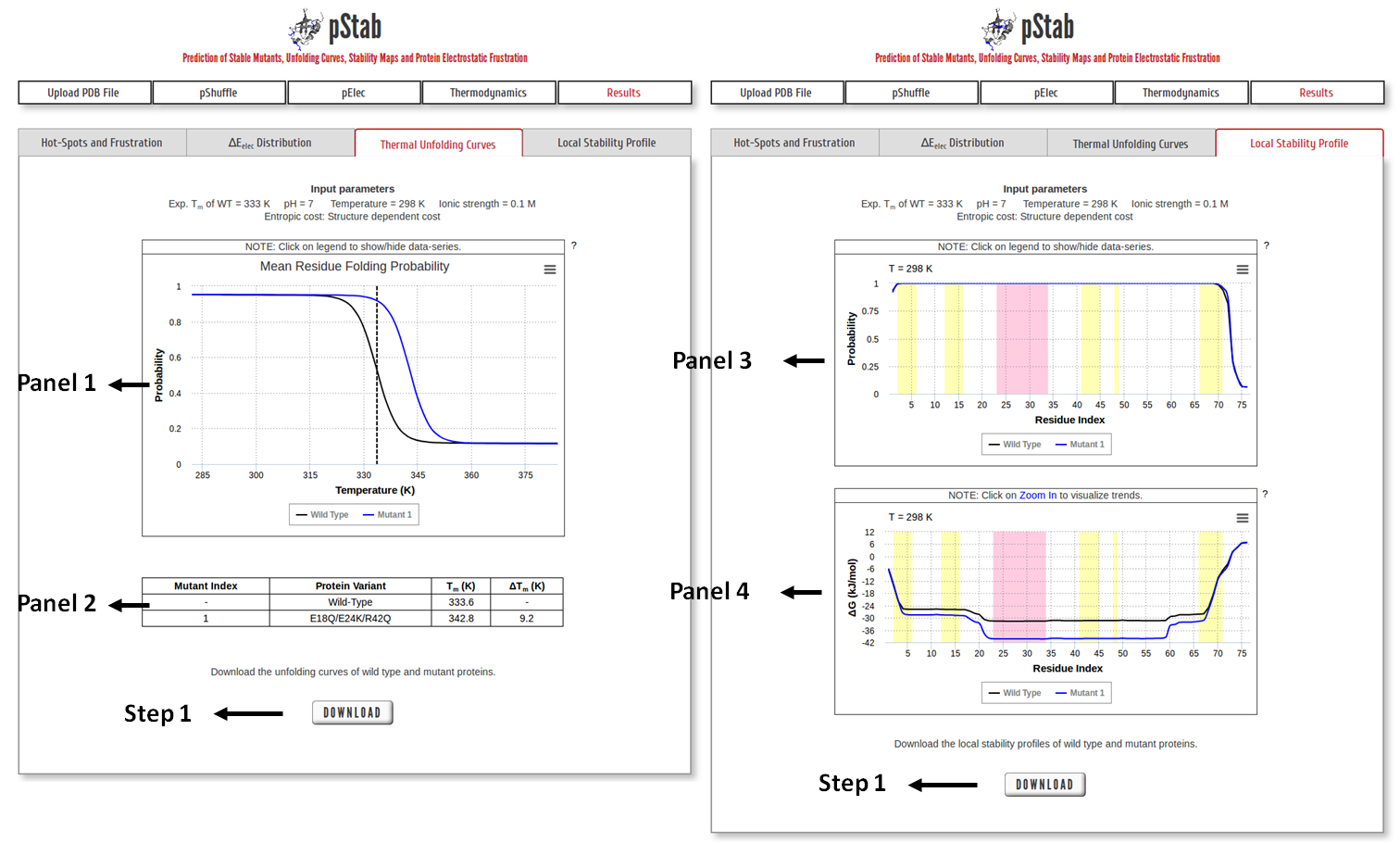
Panel 1: Thermal unfolding profile of WT protein and mutant as a function of temperature (in Panel 1), along with the details of their respective Tm (in Panel 2). Vertical dashed-lines mark the melting temperature of the WT protein.
Panel 3: Residue Folding Probability as a function of residue index. The shaded regions represent the secondary structure elements in the input PDB file (magenta: α-helix and yellow: β-sheet).
Panel 4: Same as panel 3 but for local stability (∆Glocal) as a function of residue index.
Step 1: Click on “DOWNLOAD” to save the corresponding data files into your local machine.
Electrostatic Potential Energy
The net electrostatic interaction energy (Eelec) is modeled employing the simplified Debye-Hückel (DH) formalism as described by the equation below,
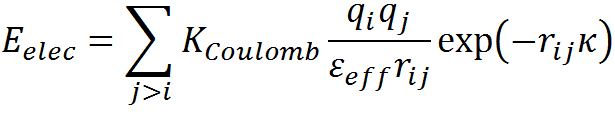
Here, KCoulomb is the Coulomb constant (1389 kJ.Å/mol), qi is the charge on the atom i, rij is the distance between charge centers i and j, and εeff is the effective dielectric constant. The magnitude of the effective dielectric constant is fixed to 29 from previous calibrations that involved comparing four different homologous protein families (Naganathan AN, 2012), 138 single point mutations involving charged residues (Naganathan AN, 2013) ) and also through comparisons of mesophile-thermophile protein pairs. 1/κ is the Debye screening length that depends on εeff, solvent ionic-strength (I) and temperature (T) as,
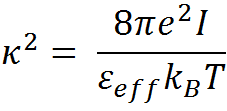
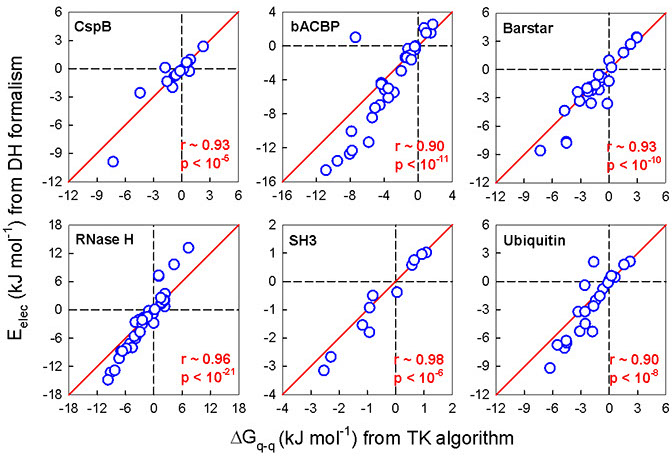
The simplified electrostatic potential energy function in combination with the carefully calibrated effective dielectric constant value (εeff) performs as well as the more complex Tanford−Kirkwood (TK) algorithm.
Structure-Based Statistical Mechanical Model
To predict the folding thermodynamics of the proteins at a residue-level resolution, we employ an extended version of the simple yet detailed Wako-Saitô-Muñoz-Eaton model. The WSME model incorporates multiple energetic terms like vdW interactions, electrostatics, solvation, and residue specific conformational entropy in its energy function. In this model, residues are assumed to sample two sets of conformations: folded (native) and unfolded (non-native) that are assigned the binary variables 1 and 0, respectively thus, leading to a total of 2N structured microstates or conformations for an N-residue protein. In the current version of model, the free energy of each microstate is
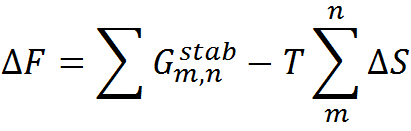
where the effective stabilization free-energy of a particular microstate (m, n) (i.e., a string of 1’s between and including m and n) at a temperature T is represented as a sum of van der Waals interactions (EVdW), electrostatic potential (EElec) and solvation free energy (∆Gsolv). ∆S accounts for the entropic cost or penalty for fixing a residue in the native conformation. The details on the model parameters and the values assigned to them by the server can be found here. Further details on the model and equations to calculate the folding probability of residues can be found in the literature (Wako H & Saitô N, 1978; Muñoz V & Eaton WA, 1999; Naganathan AN, 2012).
Errors and Warnings
| S.No. | Error | Reason |
|---|---|---|
| 1 | The PDB file does not contain protein molecule(s). | The file uploaded is either empty or does not include information on at least one protein molecule. |
| 2 | PDB ID does not exist. Please enter a valid chain ID. | The user has entered an invalid PDB ID. |
| 3 | The PDB file contains non-standard (or) post-translationally modified amino acid(s). | The pShuffle and Thermodynamics modules do not account for potential stability effects due to non-standard amino acids. |
| 4 | The PDB file contains only CA (Cα) atoms. Please upload a PDB file with complete side-chain description. | The server predictions are structure-based and hence require the complete atomic description of every residue. |
| 5 | The protein length is not within the limits (30-300 amino acids) accepted by the web-server. | The server generates mutant by combinatorially mutating the charged and large polar residues. Beyond this limit the number of possible mutants become astronomical (~ >1010) and thus intractable. |
| 6 | The PDB file is not complete. | The uploaded protein molecule has residues missing. |
| 7 | The PDB file contains multiple amino acids at the same residue position. | Multiple amino acid entries are detected with same residue number. |
| S.No. | Warning | Reason |
| 1 | For this protein, we can generate only the electrostatic frustration map and ΔEelec distribution. The protein length should be ≤ 150 residues for predicting unfolding curves and local stability maps. | The WSME model, in the cuurent version, does not account for the complex energetic description required for modelling large proteins (see references). |
| 2 | Considering only first NMR model from the uploaded PDB file. | The atomic description from the first NMR model is alone considered for predictions. |
| 3 | The PDB file contains multiple peptide chains; considering only the first chain. | If user does not provide the chain ID, first protein chain from the uploaded file will be considered for further predictions. |
| 4 | Residues with missing atomic description are modeled using PyMOL rotamer library. | The residues with missing atoms are modeled using the PyMOL rotamer library. Please verify the fixed pdb file before proceeding with the predictions. Try WHAT IF web server for more detailed modeling of the missing atoms. Caution is required when the backbone atoms are missing. |
| 5 | The PDB file contains multiple entries for the same atom(s), considering the first atomic description for each instance. | The first instance of the atomic description is considered for each atom, while the other entries are ignored. |
| 6 | Ignoring ligand(s)/hetero-atom(s) in the uploaded PDB file. | Ligand(s) and other hetero atoms present in the PDB file are ignored during the server prediction. |
| 7 | The PDB file contains disulfide bond(s). | The effects of disulfide bonds are not accounted for in the current version of the web-server. |
Further Reading
1. Wako, H.; Saitô, N. Statistical Mechanical Theory of Protein Conformation 0.2. Folding Pathway for Protein. J. Phys. Soc. Jpn. 1978, 44, 1939−1945. ![]()
2. Muñoz, V.; Eaton, W. A. A Simple Model for Calculating the Kinetics of Protein Folding from Three-Dimensional Structures. Proc. Natl. Acad. Sci. U. S. A. 1999, 96, 11311−11316. ![]()
3. Naganathan, A.N. Predictions from an Ising-Like Statistical Mechanical Model on the Dynamic and Thermodynamic Effects of Protein Surface Electrostatics. J. Chem. Theory Comput. 2012, 8, 4646-4656. ![]()
4. Naganathan, A. N. A Rapid, Ensemble and Free Energy Based Method for Engineering Protein Stabilities. J. Phys. Chem. B. 2013, 117, 4956−4964. ![]()
5. Rajasekaran, N.; Gopi, S., Narayan, A.; Naganathan, A. N. Quantifying Protein Disorder through Measures of Excess Conformational Entropy. J. Phys. Chem. B. 2016, 120(19), 4341-4350. ![]()

pStab
Prediction of Stable Mutants, Unfolding Curves, Stability Maps and Protein Electrostatic Frustration
Protein Biophysics Lab
How to use pStab
To generate mutants and analyze their effects on stability, please follow the steps below

2017, Maintained by Protein Biophysics Lab, IIT Madras, Chennai-36, India
Last Updated: September 13, 2017